

有機材料的性質:密度、內聚能、汽化熱
本案例介紹一系列直鏈烴液相的密度、內聚能密度(CED)與氣化焓的計算,包括了建模、模擬與分析方法的綜述。有機分子和聚合物體系的原子級別的模擬結果的準確性顯示,使用PCFF+力場計算得到的密度和氣化熱的平均絕對誤差為0.23%和0.28%。
關鍵詞:MedeA?環境、LAMMPS、內聚能密度(CED)、烴、密度、力場、PCFF+、COMPASS、分子動力學(MD)。
背景
有機材料的各種形式在工程上非常重要,因此,能夠準確的預測有機材料的各種性質一直受到相當的關注。其中,簡單烴類化合物作為溶劑、燃料等的應用非常重要,實驗數據也比較完善,因此特別適合用于驗證模擬方法的可靠性。模擬技術經驗表明,烴類化合物的模擬精確度在一定程度上也代表了任意的有機體系的精確度。
原子級別的分子動力學計算的原理是在計算各原子的受力后,對牛頓方程進行積分,從而得到體系性質隨時間變化的過程。用于計算體系能量和力的函數形式稱為力場[1],力場的精度決定了性質模擬的精確度。采用合適的力場,分子動力學模擬就能夠得到高精度的結果[2-6],如本文所示,模擬與實驗的結果吻合度相當高,常在1% 以內。現代的有機物力場幾乎覆蓋了所有常見分子體系,并具有都具有高度的適用性。
本文以直鏈烴化合物為例,詳細介紹了的有機液體模型構建、分子動力學計算過程和詳細的結果分析。
研究使用的程序
模擬研究使用MedeA?平臺中包含的LAMMPS [7](即由Sandia National Laboratories開發的“Large-scale Atomic/Molecular Massively Parallel Simulator”)完成。LAMMPS在多處理器的服務器上具有高度優化的分子動力學并行計算效率。 MedeA? 平臺為LAMMPS提供了靈活的使用環境,可以用來快速構建模型、設置力場參數、設計模擬流程、管理和分析計算結果。
構建分子模型
進行分子動力學模擬,首先需要創建初始的結構模型。烴類化合物的初始結構可以用MedeA?Polymer Builder創建,如圖1。MedeA?Polymer Builder可以從標準重復單元庫構建聚合物模型,可以處理各種組分和立體化學。使用者還可以根據需要構建任意的重復單元,并保存在結構數據庫中以供使用。
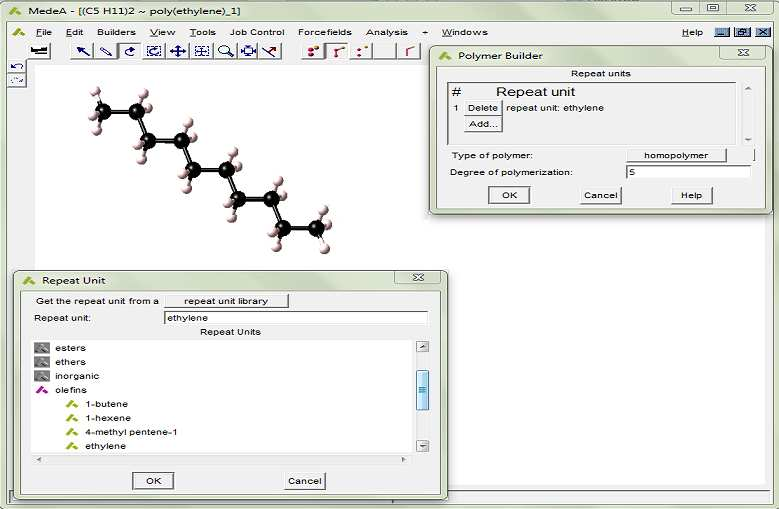
圖1. MedeA? Polymer Builder可以快速構建各種復雜的寡聚物和聚合物
從圖1得到的分子結構出發,可以很容易創建各種多分子模型。例如,用超胞構建工具創建簡單的分子重復體系,之后用分子動力學方法使體系達到液體密度的平衡態(見圖2)。凝聚態高分子模型還可以用MedeA? Amorphous Cell Builder [8]。進一步的分子動力學計算可以給出液體的物理和能量性質。
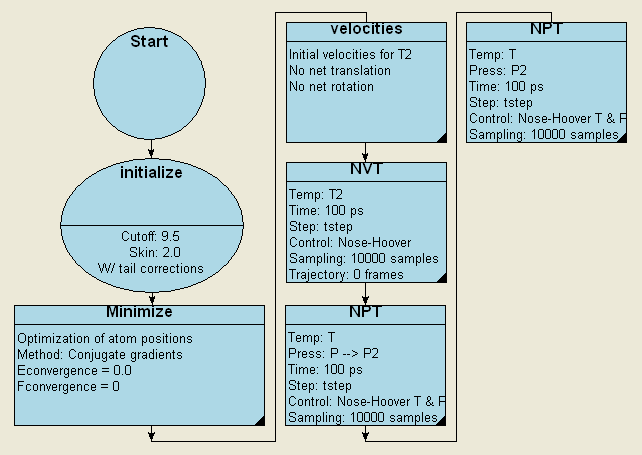
圖2. MedeA? LAMMPS 模擬凝聚相烴類化合物的流程圖
第一個NPT步驟在100 ps時間里外壓從高壓減小至設定壓力,第二個NPT步驟則將壓力恒定在P2 = 1 atm下
選擇力場
MedeA?為LAMMPS使用的力場提供了靈活的支持。力場原子類型可以根據標準模板自動指定。MedeA?提供了多種廣泛使用的力場。本研究使用的PCFF+力場是在PCFF力場[2]基礎上進行精修,尤其是非鍵參數部分是Materials Design的研發團隊的成果,所用方法與早期開發的COMPASS力場[3-6] 類似。
模擬過程
模擬過程可以概括如下:
用MedeA? Polymer Builder構造正己烷模型,用MedeA? LAMMPS進行NPT分子動力學計算。
評估性質計算對模型大小和模擬時長的敏感性。確定最佳模擬體系大小和時長。
使用相同方法研究從正戊烷到正二十烷所有直鏈烴。
選擇計算參數
圖3給出了正己烷密度隨體系大小(a)和模擬時長(b)的變化關系,不確定度由模擬過程的漲落給出。很明顯,對于正己烷來說,包含216個分子的模型和200ps的時長可以很好的描述體系的密度。經驗表明,對于絕大多數體系來說,3500個原子的體系大小與200ps的NPT時長可以給出足夠好的結果。實驗上正己烷在298.15K下的密度是0.6548g/cm3,模擬結果得到0.6547g/cm3,模擬值僅比實驗值低0.02%。
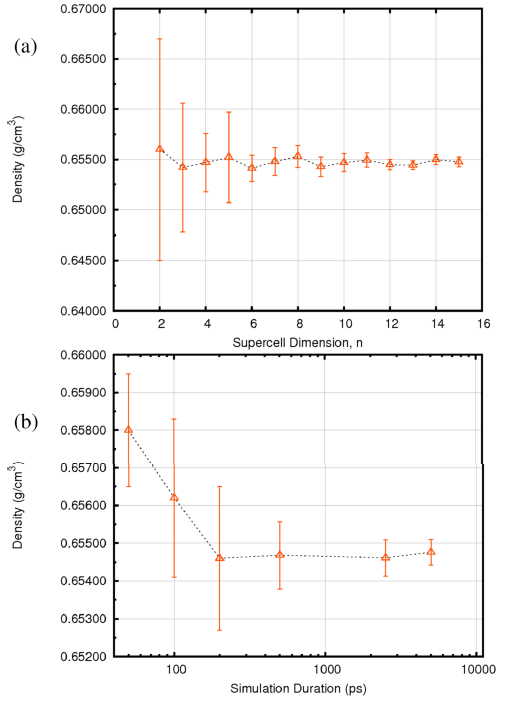
圖3. 正己烷密度的計算結果(25°C,1 atm)隨(a)體系大小(分子數為n3)(b)模擬時長(ps)
對于正己烷,216個分子(n=6)、200ps時長即可得到收斂結果
計算烴類性質
MedeA? LAMMPS的流程圖方法可以用于任何體系,將流程圖保存下來,即可重復使用,這便于將一套模擬過程應用與不同的體系。計算得到的性質結果見表1。 PCFF+給出的密度相對實驗值的偏差在0.5%以內;表1中也列出了內聚能密度(CED),實驗上的氣化熱與內聚能密度密切相關,使用PCFF+力場的計算數據與實驗值偏差小于1%。PCFF+給出密度和氣化熱的平均絕對誤差分別為0.23%和0.28%,使用PCFF+比COMPASS力場得到了更準確的結果。
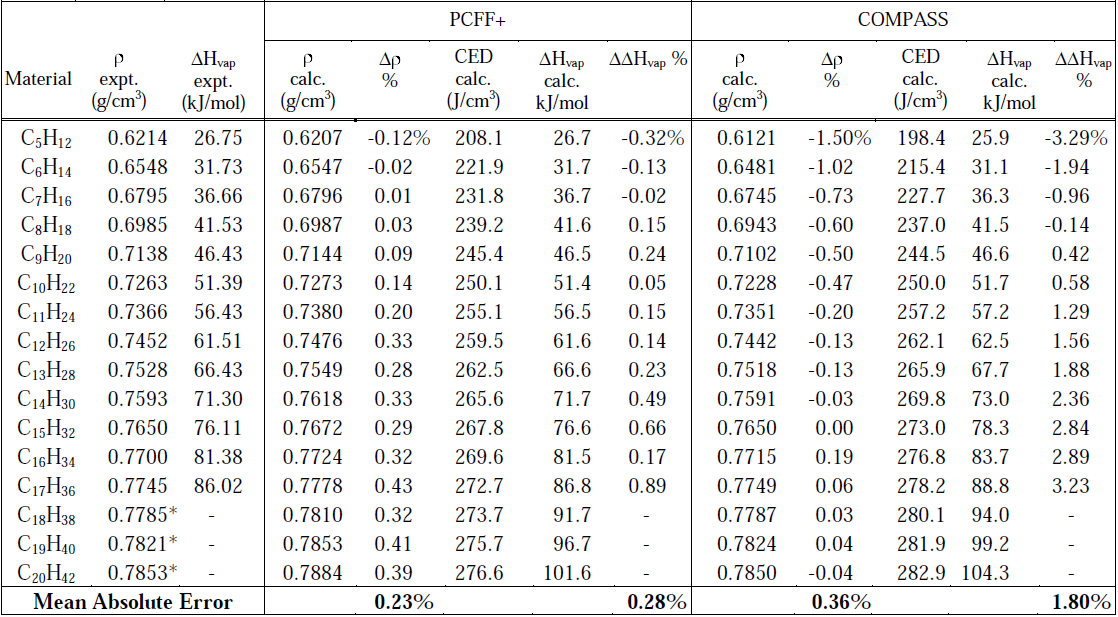
表1. 計算得到的烷烴在25°C、1atm下的密度(ρ)、內聚能密度和氣化熱。D ρ%和DDHvap%為實驗和計算值的偏差。內聚能密度由經過Materials Design修改過的LAMMPS [11]給出,密度和氣化熱的實驗數值從參考文獻[11]和[12]給出。*低于冰點的過冷液體的外推值。
其他模擬的方向
表1的結果顯示烴類化合物的性質計算可以得到與實驗數值吻合的密度、內聚能密度和氣化熱。很多其他性質,例如,熱導率、粘度、密度隨壓力的變化函數等也可以得到,而且模擬得到的結果與實驗符合得很好。此外,小分子(少于15個碳原子)擴散性質也可以用類似方法得到,相應的自擴散系數可以直接用分子動力學方法[13]給出。分子模擬的在材料性質預測上的成功使得在實驗上合成各種材料之前進行計算虛擬篩選就成為可能。
采用分子動力學進行材料性質模擬也有局限性。分子動力學過程軌跡通常只有納秒尺度,因此模擬緩慢的動態過程,例如高分子體系,大分子的擴散等,難度很大。研究體系的大小也通常限制在幾千個原子之內。
突破這種限制的的關鍵在于計算性能的提升。由于LAMMPS程序可以很方便的進行大規模的并行計算,隨著超級計算機的發展,分子動力學模擬的時間尺度逐漸進入微秒級別[14],體系大小也進入上百萬個乃至幾十億個原子[15, 16]。只要采用合理的方法和足夠的計算資源,分子動力學在預測與研究有機材料的性質等方面將會起到越來越重要的作用。

圖4. 分子動力學過程的一個快照。正己烷,25°C ,1atm
研究中使用MedeA?模塊
本計算研究使用MedeA? 材料模擬與設計平臺完成,使用了集成在其中的多個模塊:
MedeA? 平臺環境
MedeA? 聚合物建模工具
MedeA? LAMMPS接口
MedeA? Job Server
MedeA? Task Server
參考文件與注釋
基于早期分子振動性質得到的經驗能量函數和參數的計算通常使用“力場”這個名詞來描述函數形式和參數。
H. Sun, S. J. Mumby, J.R. Maple, A. T. Hagler, J. Phys. Chem. 99, 5873 (1995)
H. Sun and D. Rigby, Spectrochimica Acta A153, 1301 (1997)
D. Rigby, H. Sun, B.E. Eichinger, Polymer International 44, 311 (1997)
H. Sun, J. Phys. Chem. B102, 7338 (1998)
H. Sun, P. Ren, J.R. Fried, Comput. Theor. Polymer Sci. 8, 229 (1998)
S. Plimpton J. Comp. Phys. 117, 1 (1995); LAMMPS網站: http://lammps.sandia.gov
The MedeA? Amorphous Cell Builder 2012年發布.
力場分配模板規則使用化學結構語言來識別原子團,并分配相應力場的原子類型。原子部分電荷也同時自動分配。
Materials Design 修改了LAMMPS 內聚能密度的計算。這部分的源代碼可以向作者索取。表1中的數據通過對1000個分子構成的元胞進行500ps模擬得到軌跡數據中獲得。
F.D. Rossini, Selected Values of Physical and Thermodynamic Properties of Hydrocarbons and Related Compounds, Carnegie Press, Pittsburgh (1953)
Enthalpies of Vaporization of Organic Compounds: A Critical Review and Data Compilation, Blackwell Scientific Publications, Oxford (1985)
Y. Iwai, H. Higashi, H. Uchida, Y. Arai, Fluid Phase Equilibria 127, 251 (1997)
M.L. Klein, W. Shinoda, Science 321, 798 (2008)
M. Buehler, A. Hartmaier, M. Duchaineau, F.F. Abraham and H. Gao Acta Mech Sinica 21, 103 (2005)
F. Abraham, R. Walkup, H. Gao, M. Duchaineau, T. D. De La Rubia, M. Seager Proc. Natl. Acad. Sci. 99, 5783 (2002)