服務


ONETEP
最新版本: 無版本號
ONETEP是針對于大體系計算的具有革命性的基于量子力學的程序。ONETEP在密度矩陣公式中使用密度泛函理論(DFT)。在ONETEP中,密度矩陣是根據特殊的最大局域泛函和非正交的廣義萬尼爾函數得到的。ONETEP是一個線性標度的方法,所以隨著體系原子數目的增加,計算總能的時間是線性增加的。使用ONETEP進行第一性原理量化計算的典型應用有表面化學,結構性質,大分子體系的構象研究以及碳納米管的結構和能量計算。同樣也可以研究半導體和陶瓷材料中缺陷的相關性質(空位,空隙,摻雜,晶界和位錯)。
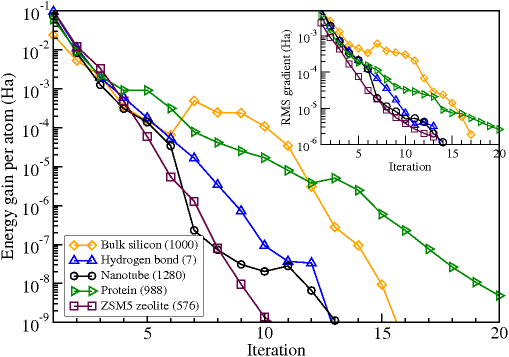
使用ONETEP程序中NGWF方法優化不同體系的能量收斂情況,括號中的數字表示體系中的原子數。
FPLO
最新版本: 9.0
FPLO是完全勢局域軌道最小基代碼,用局域自旋密度近似(LSDA)求解規則格子的Kohn-Sham方程。可以進行四分量相對論求解,以及進行LSDA+U計算。雖然程序的基組小了一個數量級,但是可以獲得與完全勢LAPW接近的數值精度。特別是計算的總能量絕對值與WIEN 97的符合程度在1 mHartree以內。由于FPLO使用了最小基,因此它可以對包含一百個過渡金屬原子的晶胞進行高精度的完全勢計算。FPLO包含圖形界面的輸入文件編輯器,3D結構工具和結果顯示工具。
FPLO-9的新功能:
1. 執行更快(最高達一個數量級),特別是對大晶胞和重元素
2. GGA加入PBE
3. LDA/GGA、LSDA+U/GGA+U實現了用力來優化內部參數(Wyckoff位置)
4. 在后期處理中可以使用Wannier函數
5. 有限尺寸核模型
6. 輸出格點的密度、自旋密度和勢,用于顯示
USPEX—新材料和礦物相晶體結構預測軟件
對在高溫、高壓等極端環境中材料結構的變化,以及發現材料新的物相是目前材料學研究領域的熱點和難點。USPEX對這一難點問題取得了突破性的進展。USPEX是Universal Structure Predictor: Evolutionary Xtallography的縮寫,由Artem R. Oganov研究小組開發的一種計算方法和同名軟件實現。她克服了使用傳統方法中遇到的成功率低和計算成本高的缺點,成功地實現了對于任意給定溫度、壓強條件下, 無需實驗數據等經驗參數,僅從材料化學成分組成進行晶體結構預測的功能。“uspekh” 在俄語中是“成功”的意思,也顯示了這種方法近100%的成功率!
特色功能
◆無需實驗數據,僅從材料的化學成分出發預測晶體結構,特別適用于高溫、高壓等極限條件下的晶體結構和分子結構預測。
◆功能性材料,如超硬、超密材料,高/低k介電材料等
◆新能源材料,如儲氫材料等
◆金屬,超導體,金屬有機物等材料
◆支持各種晶胞結構的搜索。
◆由實驗得到的晶胞結構開始搜索,如晶胞參數,晶胞形狀,晶胞體積等
◆由已知和假設結構開始搜索
◆高效的收斂技術。
◆遺傳進化算法(Evolutionary Algorithm)顯著地降低對非物理和冗余結構的搜索
◆微粒群優化算法(Particle Swarm Optimization)對周期性晶體結構預測
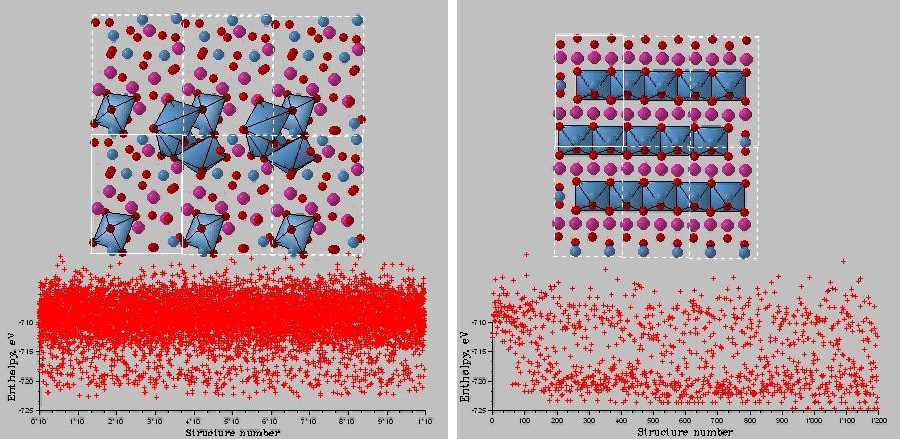
40個原子的MgSiO3超晶胞的結構搜索。左圖:結構局域優化隨機搜索;右圖:USPEX進化搜索。隨機搜索120’000步后仍未收斂,而USPEX在1’000余步即達到穩定結構。正是因為引入了進化算法中對“優質”結構的篩選和繼承,使得模擬過程快速地向低能方向收斂,同時也保留了較高能量的結構,有助于對高能區結構的探索。
◆支持分子結構的全部固定、部分固定、和完全可變的各種操作
◆使用“指紋識別”技術來確定結構的相似程度
◆支持斷點續算(可修改參數)
◆具備與強大的可視化和分析工具STM4軟件的接口
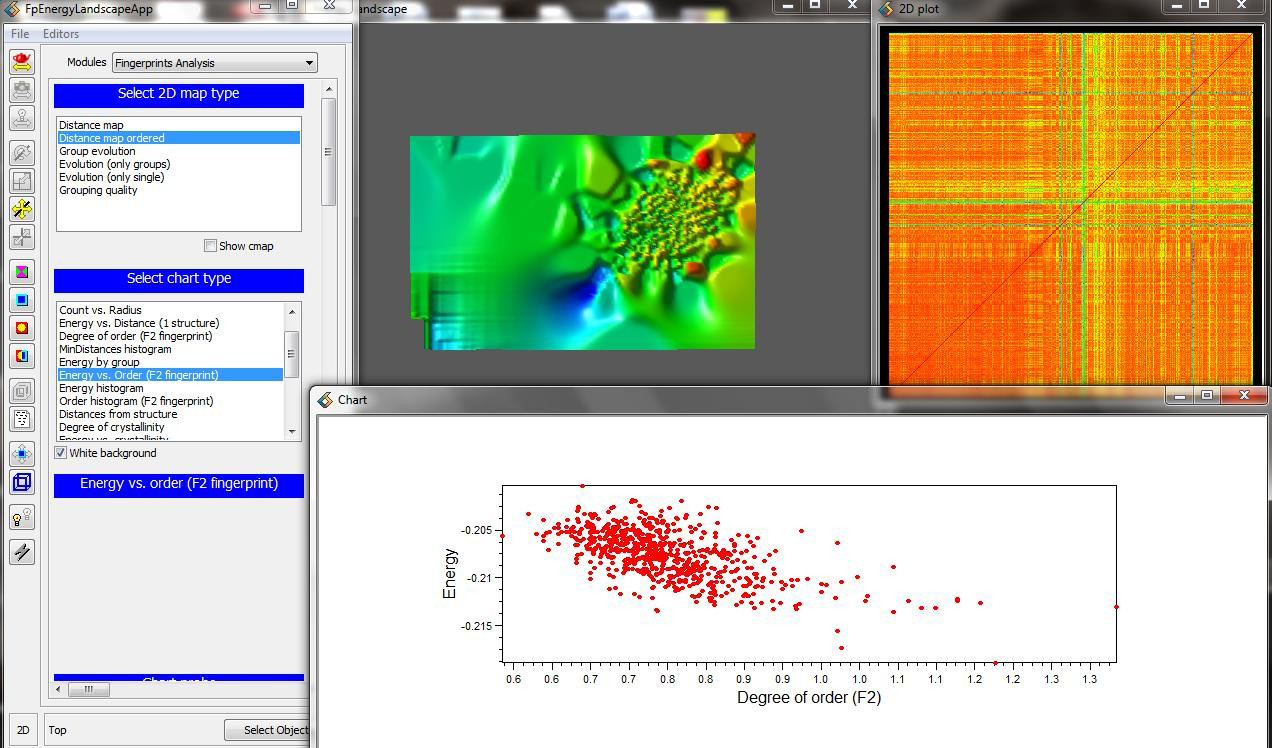
優勢
USPEX通過數片結構的空間粘連,部分保留并考慮了原子的局域排布信息。反映了晶體中強的短程相互作用和當前一代的信息。對于處理較大的體系具有明顯優勢。置換算法提供了用戶自定義哪種原子相互交換的功能,特別適用于具有長程化學相似的不同種原子構成的體系。
對于沒有任何晶體信息或者僅有晶格參數的情況下,可以完全使用從頭算處理6~40原子/晶胞的體系。對于多于40原子/晶胞的體系,計算成本顯著增大,但仍可以實現,需要借助USPEX中的其他方法或近似,足以處理大多數的晶體學和地球物理學問題。對于100~200原子/晶胞的體系,使用經典力場方法,也可以得到很好的結果。
源代碼授權
USPEX是由 Andriy O. Lyakhov, Colin W. Glass 和 Qiang Zhu 共同編寫的matlab語言的軟件包。對于大學和研究所中的個人研究者,USPEX 是開源代碼(須注冊)。對于企業用戶,USPEX 是商業軟件。北京宏劍公司是USPEX中國區合作伙伴,向國內用戶提供USPEX源代碼和技術支持。
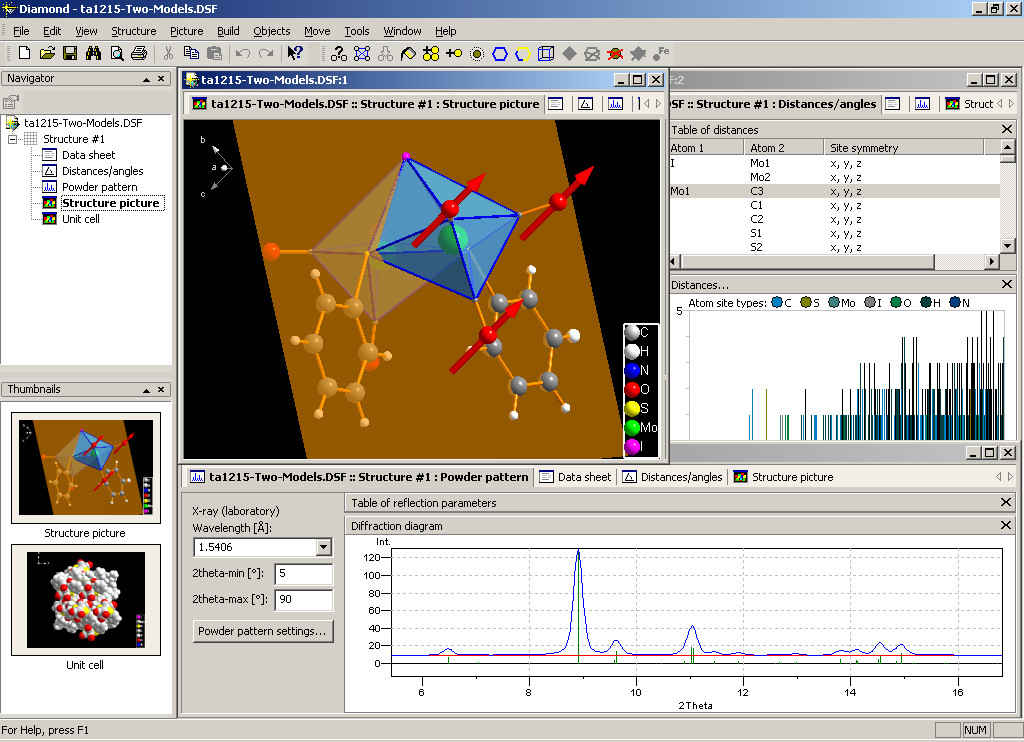
Diamond -- 晶體結構數據可視化分析軟件
最新版本: 4.5.2
Diamond是杰出的分子和晶體結構數據可視化分析軟件。它整合了豐富的功能,可以用于含有晶體結構數據的工作,適用于教育,科研以及出版。Diamond像其它的軟件一樣不僅可以畫出精密的分子和晶體結構圖片,它還有一系列拓展的功能,它可以讓你很容易的從一套基本結構參數(晶胞,空間群和原子的位置)中模擬任意部分的晶體結構。
更新功能:
WIEN2k
作為專業的第一原理周期性材料計算引擎,WIEN2k軟件包是目前采用密度泛函理論(DFT)計算周期性體系電子結構的最精確的計算程序之一。可以對各類周期性體相和表面材料實現高精度的性質計算和模擬。
可視化圖形界面:WIEN2k軟件包可提供友好的網頁終端操作環境(w2web),實現簡潔明快的作業生成、修改、遞交和管理。
WIEN2k軟件性能
最精確的全勢(Full Potential, FP)方法結合(線性)增廣平面波((L)APW-局域軌道(lo)基組和四面體布點方案,完美實現高精度計算
- 可對價層電子可考慮標量相對論效應和旋-軌耦合效應
- 對芯層電子,在原子結構層面上考慮了完全相對論的效應
- 交換-相關能的計算主要采用局域(自旋)密度近似(L(S)DA)或廣義梯度近似(GGA)
應用四面體布點方案,保證了對金屬和其它導體材料的Fermi面實現精準計算。
針對含有d或f電子的重元素體系,WIEN2K程序包可通過LDA/GGA+U方案校正局域電子的自相關作用,得到與實驗吻合的計算結果。
方便多樣的并行計算模式(k點并行、mpi并行和混合并行),大大提高了計算效率。
計算模塊及功能
內置230個空間群列表,(輔助XCrysDen)可方便地實現輸入結構的圖形化
常規的電子結構自洽迭代(SCF)計算
周期性體系的態密度和能帶結構計算
磁性體系自旋極化計算,精確計算鐵磁性和反鐵磁性結構計算和設計
X射線發射和吸收譜計算
簡單和復雜體系的結構優化,包括晶胞參數和原子內坐標 (兩者尚不能實現同時優化)
固體的光學特性和電子能量損失譜計算
超晶胞生成模塊,模擬界面和表面材料、復雜的摻雜體系
LDA/GGA+U實現含有稀土元素等重元素材料化合物材料的高精度的性質計算
WIEN2k程序包計算周期性體系舉例
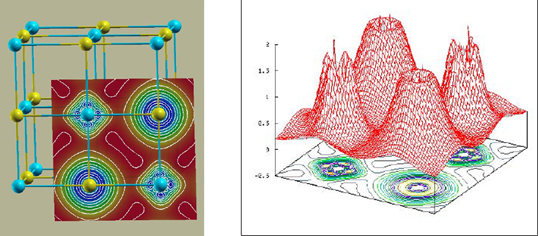
TiC的電荷密的二維剖面圖 [(100)面]和三維等值線圖
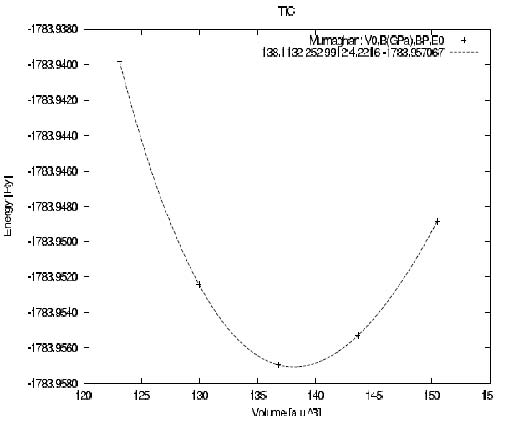
周期材料結構TiCd的晶胞參數的優化
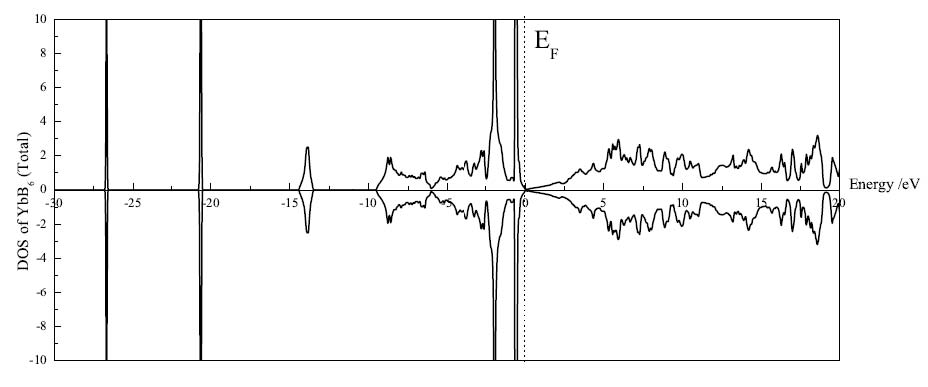
摻雜體系CaxEu1-xB6自旋極化態密度(對Eu的4f電子考慮GGA+U)
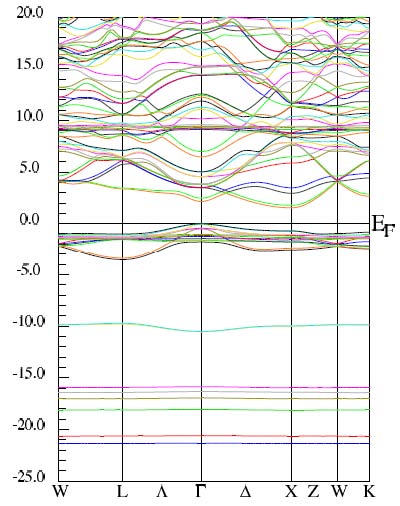
考慮旋-軌耦合后EuTe的能帶結構(對Eu的4f電子考慮GGA+U)。
<-15.0eV區域,考慮旋-軌耦合后Eu的5p1/2與5p3/2軌道顯著裂分。