


第一輪通知
尊敬的老師:
在“第七屆國際理論化學、分子模擬和生命科學研討會暨科學計算和模擬軟件發展平臺學術交流會”(7月21日-23日)結束后,將于7月24日,在北京師范大學隨后舉辦“基于GPU計算的分子動力學軟件及量子化學計算軟件學研班”。
本學研班將分別介紹基于GPU計算的分子動力學軟件-ACEMD、TeraChem軟件(暢享高速高效并行GPU平臺),從頭算量子化學程序包Q-Chem、Molpro及Molcas軟件的理論及應用,學習及研討有關分子動力學模擬和分子力場的基本原理、基本方法和相關重要程序。授課專家為相關領域的知名學者,其中也有所講授軟件的開發者。授課專家結合個人的科研經驗和實例為大家介紹基本理論、方法及軟件的使用。同時,北京宏劍公司根據廣大用戶的實際需要,幫助學員解決如何將程序用于科研的實際問題。學習和研討內容亦多結合當前有關分子動力學模擬和分子力場、量子化學計算軟件的前沿科研課題。
2 主辦單位:北京師范大學
2 支持單位:北京宏劍公司 Global Simulation
2 授課專家:于建國教授 北京師范大學;劉俊博士 北京宏劍公司
2 培訓時間:2013年7月24 日
2 培訓地點:北京師范大學化學院 北京
2 收費標準:600元/人,參會人員住宿費及交通費自理。
2 優惠政策:參加“第七屆國際理論化學、分子模擬和生命科學研討會暨科學計算和模擬軟件發展平臺學術交流會”
注冊人員培訓費減半(300元/人)。
2 聯 系 人: 北京宏劍公司 李長瑜
2 電 話: 010-51261900-868
2 E-mail : 該Email地址已收到反垃圾郵件插件保護。要顯示它您需要在瀏覽器中啟用JavaScript。
2 會議報名網址: 報名請點擊
2 地 址:北京師范大學化學院
課程安排
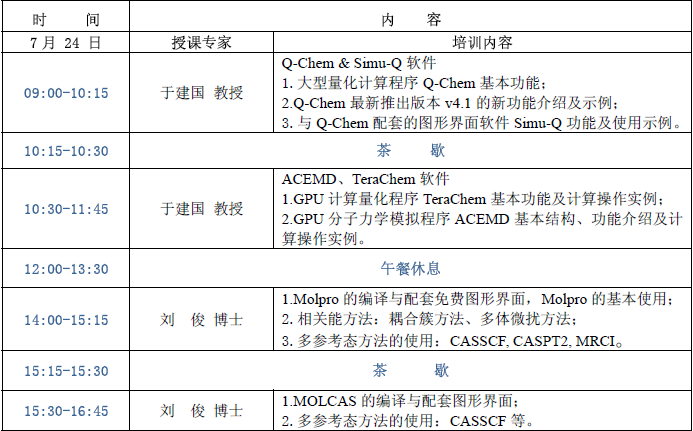
授課專家介紹
? 于建國教授:物理化學博士,北京師范大學化學學院教授,長期從事理論與計算化學基礎研究與教學,為自主知識產權的計算化學模擬軟件SimuPac的設計及開發者,國際著名計算化學軟件
Q-Chem、Spartan和AMPAC的作者之一。
? 劉 俊博士:物理化學專業,北京宏劍公司高級技術支持,進修于英國 Cardiff大學并接受Peter Knowles教授培訓,掌握Molpro最新版本,具備培訓師資質。主要從事相對論量子化學的理論研究和程序
化,對密度泛函等理論、從頭算理論與方法有比較深刻的理解,尤其是對兩種Kohn-Sham 有效勢進行了深入的研究。將OEP理論應用于精確的4分量相對論與2分量相對論中,提出SAOP的
局限性與解決。熟悉量子化學程序編寫,以及各種量子化學軟件的安裝,操作和使用,計算中心軟件部署。
軟件介紹
? Q-Chem:
2 Q-Chem是一款功能齊全的從頭算量子化學程序包,全面支持從DFT/HF到各種高級的post-HF相關能計算方法,被廣泛用于處理工業界、學術界和國家實驗室的各類理論模擬研究,在理論化學、藥物設計、材料科學、生物化學以及相關領域的教學和研究中發揮了極大作用。可以包括研究分子結構、化學反應、分子振動、電子光譜、NMR譜和溶劑化效應等。
2 基于快速DFT計算的遠程校正(Dispersion)、雙雜化(double-hybrid)和按距離校正(range-corrected)的新泛函;
2 諸多用于自由基和激發態計算的EOM-CCSD方法,一系列用于強相關處理的有效活化空間方案;
2 TDDFT解析梯度計算,和目前世界獨有的TDDFT解析hessian計算,各種用于電荷遷移和激發態計算的新方案。
? ACEMD:
2 ACEMD是最領先的分子動力學GPU軟件。ACEMD是一個功能全面的的分子動力學模擬軟件包,可以高效的運行于NVIDIA GPU上。ACEMD的主要優勢是對分子動力學模擬,大大提高模擬效率(幾十甚至上百倍),提高科研效率。
2 ACEMD的主要功能及特點:
1. 全面覆蓋Amber、CHARMM、NAMD、CellMD、Gromos功能
2. 部分覆蓋LAMMPS、Gromacs的功能
3. 包含Particle mesh Ewald (PME)及廣義反應力場
4. 方便讀取Charmm、Amber等力場
5. 支持metadynamics, steered MD等插件
6. 支持umbrella sampling, replica exchange等插件
7. 運行時間步長超過4fs
8. 剛性、柔性氫鍵
9. 完全兼容CUDA、OpenCL
10. 支持TCL腳本
11. 便于根據模擬結果不斷修改模擬條件
12. 很好的穩定性、可靠性
? TeraChem
2 世界首創在GPU上運行的量化計算程序!
2 TeraChem1.45:開拓與首創
2 TeraChem主要功能及特點:
1. 支持Nvidia的Tesla和FermiGPU的全部功能
2. RHF、UHF和DFT能量與解析梯度計算
3. 多種DFT泛函和DFT網點計算選擇
4. 幾何構型和過度態優化(包括限制性優化)
5. 從頭算分子動力學(NVE和NVT系綜)
6. 支持多GPU系統
7. 支持單精度、雙精度和混合精度計算
8. 適合超大分子計算的設計
9. 環繞水分子的QM/MM計算
10. 自然鍵軌道分析
11. 比常規的在CPU上的計算加速能達到1000倍!
? Molpro軟件
2 最新版本:2012.1
2 Molpro軟件是一款多功能全面的量子化學計算軟件,其優勢在于對電子結構的高精度計算,并且提供的算法更加全面,包括密度泛函DFT,微擾理論Mpn,多參考組態相互作用(MR-CI),耦合簇(CC)以及其他相關方法,是國際上廣泛應用的專業級量化計算軟件。尤其體現在處理激發態的時候,Molpro用MR-CI方法通常只需10個循環即可自洽收斂,而不會像其他軟件常用的CASSCF方法那樣,難以收斂,并且MR-CI的計算結果更加精確。
? Molcas軟件簡介
2 最新版本:7.8
2 Molcas軟件側重于多組態的量子化學計算,適用于研究于那些無法用單組態合理描述其他電子結構的體系,如激發態,化學反應過渡態,重元素(過渡金屬,鑭系,錒系)體系等。結合量子化學與分子的QM/MM方法可以用來計算大分子和分子簇,NEMO方法能夠為MC/MD模擬產生分子間力場。
北京宏劍公司
2013年6月21日